Preguntas frecuentes
La división celular en ocasiones no es perfecta y arrastra fragmentos o porciones de DNA de determinados cromosomas. El origen del fragmento extra en éste síndrome es en el cromosoma 15.
El cromosoma 15 es un cromosoma acrocéntrico con 106 millones de pares de bases y con unos 800 genes. El cromosoma 15 constituye el 3,5% del la totalidad del ADN humano.
Concretamente, el fragmento extra corresponde una copia de la región comprendida en el brazo largo “q” del cromosoma 15, y que se extiende desde la región q11, hasta individuos que llegan a la región q14.
El fragmento extra está formado por dos porciones de la región q11q13 que se copian de manera idéntica en el proceso de la replicación, durante la división. Éstos dos fragmentos se unen uno con el otro, pero justo antes de la unión, uno de los fragmentos realiza una inversión. En consecuencia se forma una imagen especular (en espejo) en la confrontación de ambos fragmentos. La aportación del fragmento o porción extra, solamente expresa sintomatología si es de origen es materno.
La aportación paterna en este caso, es asintomática o al menos en la bibliografía médica se desconocen casos de síndrome idic15 de origen paterno. Investigadores estadounidenses en laboratorio clínico se atreven a afirmar que cabe la posibilidad de la existencia idic15 de origen paterno que podrían llevar una vida relativamente normal ya que no han tenido ningún tipo de síntoma.
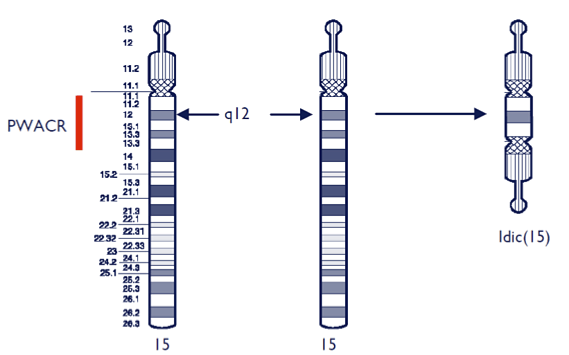
El diagnóstico del síndrome se confirma con un estudio cromosómico “cariotipo” en el que puede observar un cromosoma número 15 extra. El cariotipo pone de manifiesto la triplicación del cromosoma implicado, el que el individuo afectado posee una trisomía en el par 15.
El cariotipo nos da una información de tipo numérico y estructural. Pero si realmente, queremos tener la información de la estructura a nivel de regiones en la secuencia del DNA es más específico un estudio molecular, un estudio de hibridación por fluorescencia in situ ó “FISH”. Éste estudio indica el tamaño del fragmento de la región duplicada.
Existe además otra técnica de hibridación por fluorescencia que nos permite detectar variaciones en el número de cromosomas, en el que se si existe pérdida o ganancia de cromosomas completos, ésta técnica se denomina “CGH-array”
Resultados
La manera de expresar el resultado del cariotipo sigue unas normas establecidas pero podemos decir en un mismo resultado que es un derivado del 15, como que es una inversión duplicación, como que es un isodicétrico. En los casos de idic15 intersticiales la formulación es la misma pero con 46 cromosomas. En los casos de mosaicismo se ponen porcentajes de afectación numérica en cromosomas. Podemos encontrar los siguientes:
47,XX,+invdup 15(pter?q14::q14?pter).ish trp(15)(q13)(D15Z1 X 4, D15S10 X 4)
47,XY,+mar.ish dic(15)(D15Z1++,D15S10++,SNRPN++)
47,XY+der(15)(q13;p11.2)(pter->q13::p11.2- >pter)
46, XX, + mar.ish idic (15)(q11q13)
47,XX+ der (15) inv (15)(q11;p11.2) [40%] / 46,XX [60%]
47,XX,idic(15)(q11)dn
47,XX,+psu dic(15)(q11q13)
46,XX,+inv dup(15)(q11q13)
47,XY.ish idic(15)(q13)(D15Z1x2,SNRPNx2,PML-)
Hay que entender que la alteración se encuentra el ADN del núcleo de la totalidad de las células del organismo (excepto mosaicos que existen %) y que no existe ningún fármaco que pueda modificar la alteración estructural ya producida. En la actualidad se están desarrollando avances el muchas enfermedades de origen genómico en las cuales se puede realizar una terapia génica (activando o inactivando genes).
La medicina en vista al futuro será una medicina predictiva y no paliativa como la actual. En el caso del síndrome idic15 se trata de paliar la sintomatología como la epilepsia, con anticonvulsivos y la hiperactividad con déficit de atención con antipsicóticos y/o sedantes. La creación de nuevos fármacos que sean capaces de activar o inhibir la expresión de los genes implicados en la región 15q11q13 son motivo de esperanza para los familiares de los afectados con una tetrasomía parcial del cromosoma 15 (idic15).
A nivel morfológico
las personas que sufren ésta alteración no tienen rasgos faciales significativos. Pero se ha observado en muchos individuos engrosamiento de los labios, nariz achatada con orificios nasales de mayor diámetro, pliegue palpebral en el canto interno del ojo “epicanto”, orejas de implantación baja con cierto grado de rotación hacia el lado posterior.
En ocasiones se observa mentón prominente en individuos de mayor edad. Los individuos no tienen porqué tener todas las morfologías faciales que se indican, bien pueden tener algunas de ellas, o incluso no tener ninguna. Por ello el fenotipo característico de una persona que posee el síndrome idi15 no sigue unos rasgos fijos.
La sospecha de ésta alteración por los rasgos morfológicos pasa totalmente desapercibida para profesionales e incluso para familiares ya que son rasgos que pueden ser considerados heredados por los progenitores.
Algunos de ellos presentan ligeros estrabismos divergentes (hacia el lado externo de la órbita).
La microcefalia es una característica que puede alertar al profesional en pediatría de un problema neurológico.
A nivel motriz
se ven claramente desfavorecidos ya que tienen alterado el tono muscular. Poseen una hipotonía muscular generalizada desde el periodo postnatal, pero se han dado casos que ocurre todo lo contrario, una hipertonía generalizada que suele mejorar con el tiempo. Por ello se habla de alteración del tono muscular de manera generalizada que les impide una coordinación de movimientos de manera fluida. Atonía muscular, deambulación atáxica. Presentan dificultades en la motricidad fina a la hora de realizar tareas con ligera dificultad como es pinchar con un tenedor una pieza troceada de fruta ó poner una moneda en la hucha.
Existe un predominio de la musculatura flexora sobre la extensora. Por ejemplo suelen caminar con la mano caída (antebrazo relajado) con el brazo flexionado (bíceps en contracción) adoptando una postura de “brazo en garza” en el brazo contrario al que le dan la mano al acompañante.
A nivel psicológico, cognitivo y comunicativo
presentan en un 90% trastorno del espectro autista (TEA), que se manifiesta por la triada característica; trastorno en la relación social, trastorno de la comunicación y conductas estereotipadas, pero que hasta los tres años de edad no se expresa con toda seguridad.
Por otro lado un 3-5% de todos los TEA de origen sindrómico corresponden al síndrome idic15. En la actualidad se catalogan como autismos idiopáticos(causa desconocida) en los casos que no se les ha realizado pruebas genéticas. De ahí la importancia del conocimiento y características del síndrome por el Neuropediatra fundamentalemente y psiquiatras infantiles.
Psicológicamente tienen un predominio hacia la impaciencia, fácilmente alterables, comunicación con predominio gestual, muy nerviosos. La capacidad de comprensión está mucho más desarrollada que la capacidad expresiva, con lo que muchos de ellos, por frustración comunicativa presentan rabietas y patadas en muchas ocasione y que suele ser motivo de preocupación para los padres en sus relaciones sociales.
Destacar aspectos psicológicos propios del TEA como la dificultad de interacción social, evitan el contacto directo, mirada huidiza, posturas extrañas, comportamiento obsesivo por determinados objetos, uso no funcional de objetos (traslado de objetos de un lugar a otro, colocación de objetos en hilera…), conductas estereotipadas, expresividad variable, cambios de humor repentinos.
La mayoría tienen fascinación por el agua y la música. Los objetos brillantes como broches, pendientes, gafas y los espejos… suelen llamar su atención. Muchos de ellos suelen tener afinidad por los animales, como las actividades con caballos por ejemplo. Sin embargo tienen impulsos obsesivos por el cabello largo y suelen estirarlos con facilidad. Además muchos de ellos si quieren solicitar atención lo demuestran sometiendo a pellizcos a las personas de su entorno.
Los signos neurológicos
más importantes desde periodo postnatal, aunque son muy variables son la dificultad para mantener cabeza erguida debida a la falta de tono muscular. La aparición de las crisis epilépticas son variables en la edad, frecuencia y tipos de crisis, incluso cambian en el mismo individuo. Bien pueden aparecer a los 3 meses con numerosas crisis al día pero de intensidad leve y duración breve, como aparecer a los 7 años de edad y ser menos frecuentes pero más agresivas. Las pruebas solicitadas para discriminar tipo de crisis y administrar una terapéutica adecuada nos la dará el resultado de un EEG.
Los estados de vigilia y sueño dentro de la alta variabilidad suelen estar alterados observables en EEG.
Poseen una hipersensibilidad a estímulos sensoriales de tipo acústico (aumentado el sistema de alerta). Ante cualquier tipo de sonido de tipo repentino e intenso suelen mostrar una respuesta excesiva. Por ejemplo un repentino aplauso, fuegos artificiales, tormentas.
Los espasmos nocturnos no se dan en todos los individuos y los que los poseen tienen una dificultad para conciliar de nuevo el sueño y las fases del sueño se ven constantemente interrumpidas, lo cual condiciona la actividad diaria.
A nivel orgánico estructural
tienen predisposición a alteraciones cardiacas que en la mayoría de casos no son graves, aunque se han dado en raras ocasiones comunicaciones entre ambos ventrículos y otras patologías más graves como “tetralogía de Fallot”.
Las anomalías en el aparato genitourinario también pueden darse, como por ejemplo hipospadias en los varones, agenesias renales e hipogonadismo. Por tal motivo se recomienda que el neuropediatra pida ecografía abdominal y ecocardiograma como pruebas complementarias para descartar posibles problemas asintomáticos.
Algunos de los afectados suelen tener paladar estrecho u ojival que suele tener su origen en un crecimiento adenoideo. Ese motivo sumado a la afectación del control voluntario de la musculatura lisa implicada en procesos de deglución, muchos de los afectados en periodo postnatal tienen procesos de atragantamiento sobretodo con los líquidos.
Algunos de los afectados muestran problemas gastrointestinales desde la infancia con episodios de regurgitación de contenido estomacal por la inervación vagal gastroesofágica.
El sistema inmunológico suele estar debilitado por lo que tienen predisposición a enfermedades infeciosas de origen estacionario como refriados comunes y gripes.
Suelen tener hiperlaxitud articular observable de manera notable en regiones distales del miembros superior concretamente en articlulaciones interfalángicas y metacarpofalángicas
- Afectación a igualdad de sexos con una prevalencia de 1:30.000
- Que todos poseen en mayor o menor grado déficit intelectual y trastorno generalizado del desarrollo (TGD).
- Alteración en el tono muscular. Hipotonía en casi todos los casos.
- Compresión y expresión limitada. Dificultades en el aprendizaje.
- Riesgo elevado a poseer TEA (90%).
- Diversidad fenotípica y clínica por la variabilidad del fragmento duplicado.